Ученые ТПУ и Китая создали универсальный метод для точного «расчета» поведения молекул
Новый метод, основанный на комбинации математических алгоритмов и данных микроволновых спектров, обещает упростить и повысить точность предсказаний спектров и динамики молекул, что важно для моделирования химических процессов на квантовом уровне и изучения атмосфер планет.

Молекулы ведут себя по‑разному в зависимости от окружения: температуры, давления, соседних частиц. Чтобы предсказать их поведение, ученым нужно знать так называемую внутримолекулярную потенциальную функцию — своего рода «энергетический ландшафт», который определяет, как атомы в молекуле движутся и взаимодействуют. Однако точно рассчитать эту функцию для сложных многоатомных молекул долгое время было крайне трудно.
Классические методы хорошо работали лишь для простейших структур. Для более сложных приходилось прибегать к громоздким численным расчетам, что снижало точность и ограничивало возможности исследователей. В итоге страдали важные прикладные задачи: от создания новых материалов до моделирования химических процессов, происходящих в атмосферах далеких планет.
Новый аналитический подход вместо численных расчетов
Команда ученых из Томского политехнического университета и Хэйлунцзянского университета (Китай) предложила элегантное решение. Они разработали аналитическую методику, которая с высокой точностью определяет фундаментальные параметры молекул на основе их микроволновых и субмиллиметровых спектров. Метод работает в широком диапазоне теории возмущений — вплоть до четвертого-шестого порядка, что позволяет учесть тонкие эффекты, ранее недоступные для анализа.
«Разработанная нами аналитическая методика позволяет с высокой точностью определять фундаментальные параметры различных молекул на основе их микроволновых и субмиллиметровых спектров. Более того, она работает в широком диапазоне — вплоть до четвертого-шестого порядка теории возмущений. Это позволяет учесть более тонкие и слабые эффекты влияния на молекулу, по сравнению с тем, что было возможно ранее. Использование нового инструмента значительно сократит объем вычислений в спектроскопии и повысит их точность», — отмечают соавторы исследования, профессора Исследовательской школы физики высокоэнергетических процессов ТПУ Олег Уленеков, Елена Бехтерева и Ольга Громова.
На основе этих алгоритмов политехники создали программы на языках программирования MAPLE и Python. Они помогают определять фундаментальные параметры молекул без скрытых допущений, которые неизбежны в традиционных подходах.
Первые успехи: тест на сероводороде
Чтобы проверить, насколько новый метод точен, ученые апробировали его на молекуле сероводорода (H₂S). Результаты полностью совпали с современными высокоточными экспериментальными данными. Это подтвердило, что методика работает и может быть масштабирована.
«Новый подход подходит для самых разных многоатомных молекул. В будущем он позволит точнее предсказывать спектры и динамику молекул как в условиях атмосфер планет Солнечной системы, так и моделировать химические процессы на квантовом уровне», — говорится в сообщении пресс‑службы ТПУ.
Исследование поддержано Министерством науки и высшего образования России (проект FSWW–2026–0046), а его результаты опубликованы в авторитетном научном журнале Chemical Physics (Q2, IF: 2,8).
В планах ученых — расширить метод на более сложные молекулы и более «высокие» параметры потенциальной поверхности, а также адаптировать его для применения к колебательным структурам молекул типа асимметричного волчка. В работе также участвовали магистранты ТПУ Сергей Сидько и Георгий Копытов, что показывает вклад молодых талантов в эту перспективную область.
Похожие статьи

Китай спустил на воду крупнейшую в мире плавучую ветроплатформу на натяжных опорах
Из порта Гаолань в Чжухае вышла в море первая в мире плавучая ветроэнергетическая платформа на натяжных опорах мощностью 16 МВт. Высота сооружения превышает 307 метров, масса — почти 8 тысяч тонн.
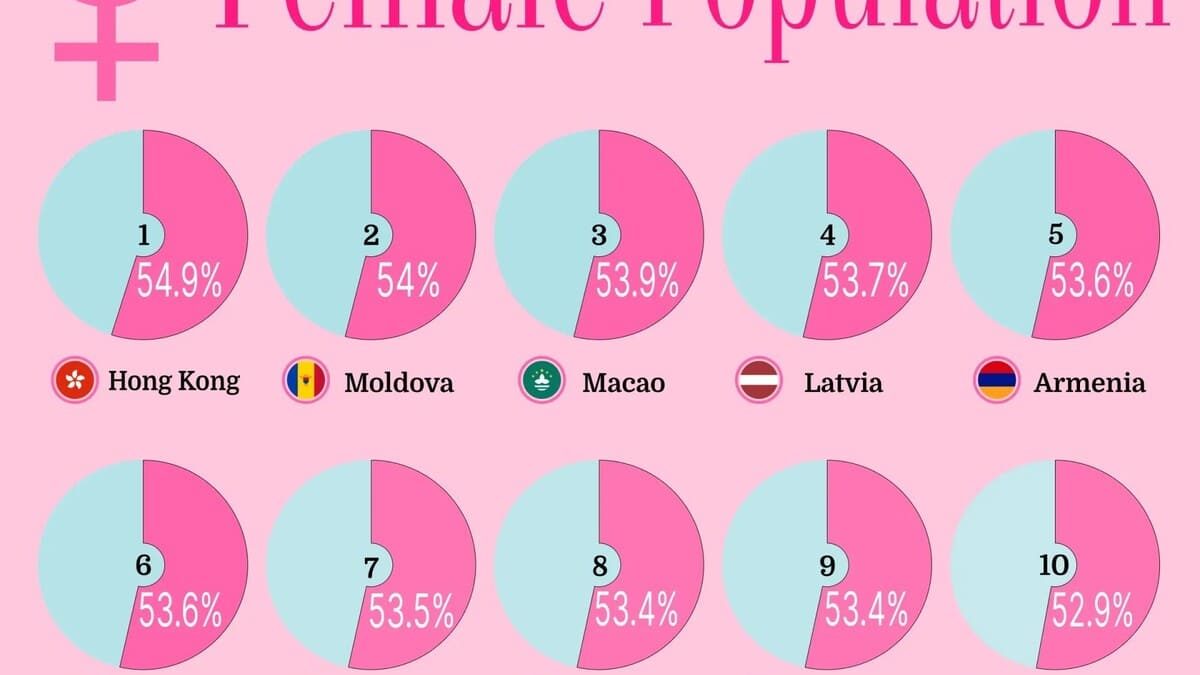
Где женщин больше: страны с наибольшим перекосом в гендерном балансе
Почти во всех странах мужчины и женщины представлены примерно поровну. Но в ряде государств и территорий женщин заметно больше — Гонконг, Молдова, Макао, Латвия и Армения входят в первую пятёрку.
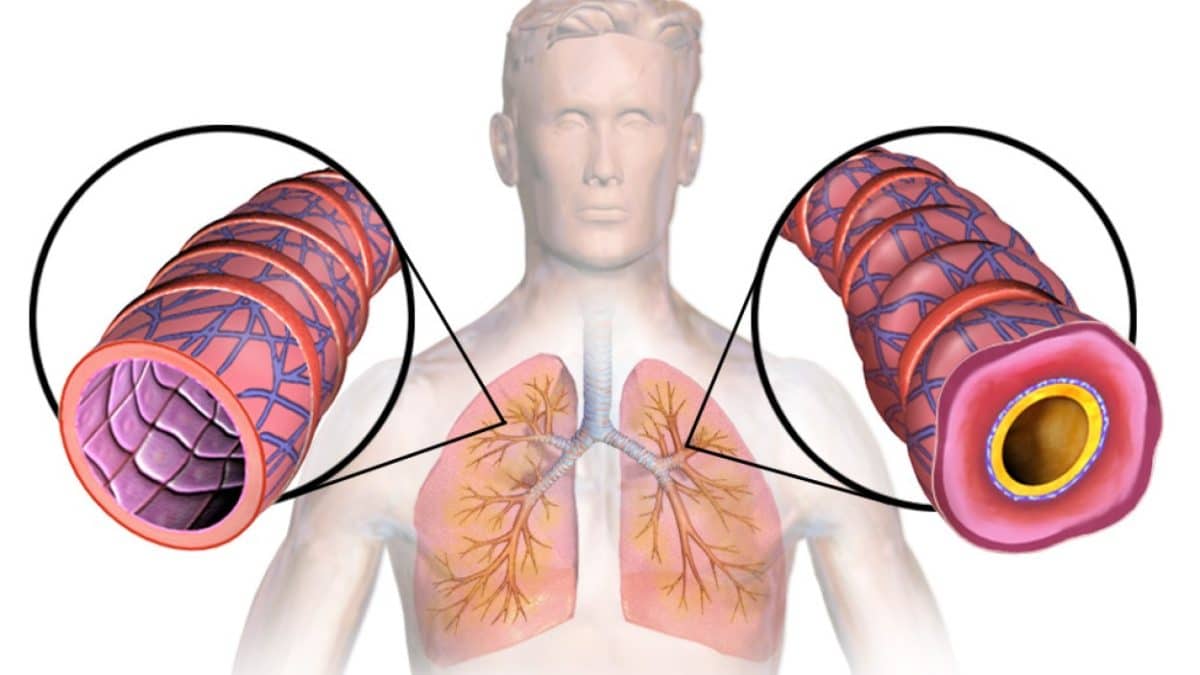
Искусственный интеллект научился различать болезни легких по выдоху: новое слово в неинвазивной диагностике
Ученые Сеченовского Университета разработали модель машинного обучения, которая по составу выдыхаемого воздуха способна дифференцировать четыре хронических заболевания легких.
Комментарии
0 всего